|
Introduction and References Quick examples Options
|
Note All above quality control functions at variants in VCF format can be turned off by --no-qc. In addition, KGGSeq can make use of phenotype information (specified by "--indiv-pheno" or "--ped-file") to filter genomic variants. This QC is quite useful when multiple samples are included in one input file, like trio or case/control study design.
Moreover, this function can work with options to flexibly set the sample size and missingness of genotypes. The following table list these options.
After running individual genotype QC, there will be two output files produced: one default Kggseq output stored in txt (or Excel by specifying "--excel"), in which the basic annotations for each clean variant are provided (see above section); the other one is clean vcf file, in which the variant row which does not meet the conditions specified will be removed completely, hence not considered in following analysis. Note The genomic variant QC is also available for non-vcf type of input, by using type of variants, or genomic position only. Sample QCKGGSeq provides two set of functions to facilitate quality assessment of sequencing data of each subject. Firstly, you can use KGGSeq to convert genotypes from VCF format to Plink binary genotype format (see Outputs part) so that routine sample QC (pairwise relatedness, mendel error rate and sex check) in genome-wide association study (GWAS) analysis can be performed quickly. The samples with unexpected relationship or error rate are suggested to be removed.
An unexpectedly high pair-wise relatedness (denoted by the PI_HAT of plink) suggests cross-individual contamination Note To estimate the relatedness (the PI_HAT), you normally do NOT need the genotypes datasets from 1000 Genome Projects (which will require rather large memory). The merging with ancestry-matched HapMap genotype data is often sufficient for an accurate estimation of relatedness. However, the estimation of PI_HAT without reference genotypes in a small local sample will result in an over-estimated value because of the inaccurate allele frequencies derived from the tiny sample. plink --bfile kggseq --mendel
plink --bfile kggseq --check-sex
Secondly, we provide plotting function (see Plotting part) to visualization the distribution of minor alleles per individual. The outlier from the distribution of total rare variants (MAF < 0.01, or novel in the common database) across all individuals can be easily detected and then ignored in the following analysis.
Note The meanings for -db-filter and --allele-freq in this section are provied in "Allele Frequency Trimming" section.
|
Filtering and PrioritizationKGGSeq offers simple but potentially powerful strategies to variants filtering and prioritization, which help users isolate most promising disease causal candiate variants from sequence variants to be considered.Users could prioritize variants based on some or all of the following evidences and/or functions: Genetic inheritance sharing, gene feature, allele frequency, biological function prediction, sequence variant type, and phenotype-relevant filtering. Genetic inheritance sharing
Filter out variants for which their genotypes are not consistent with the assumption of disease inheritance pattern. Functions and applicable models for each code of '--genotype-filter'options are listed below:
Note The inheritance mode based filtration is proposed under strong assumptions for rare Mendelian disease with clear inheritance mode. If the inheritance mode is elusive, such filtration is not suggested as it may lead to the miss of genuine causal mutation(s). This is particularly true when one has no sufficient information to distinguish the compound-heterozygosity diseases from the recessive diseases.
This function is designed for a disorder suspected to be under compound-heterozygous or recessive inheritance mode, in which both copies of a gene on the two homologous chromosomes of a patient are damaged by two different mutations or the same mutation. For recessive mode, it simply checks variants with homozygous genotypes in patients. For the compound-heterozygous mode, it can use two different input data, phased genotypes of a patient or unphased genotypes in a trio. Here the trio refers to the two parents and an offspring. When these alleles causing a disease at one locus, it follows the recessive model; and when they are at two loci, it follows the compound-heterozygosity model. In both cases, a gene is hit twice. This is the reason why it has the name 'double-hit gene'. Besides the file storing prioritized sequence variants, this analysis with --double-hit-gene-trio-filter option will lead to a list of double-hit genes in at least one subject. The counts of double-hit genes of each subject are saved in *.doublehit.gene.trios.flt.count.xls (or *.doublehit.gene.trios.flt.count.txt) and the genotypes of involved sequence variants are saved in *.doublehit.gene.trios.flt.gty.xls (or *.doublehit.gene.trios.flt.gty.txt). The corresponding files produced by the analysis with --double-hit-gene-phased-filter option are *.doublehit.gene.phased.flt.count.xls (or *.doublehit.gene.phased.flt.count.txt) and *.doublehit.gene.phased.flt.gty.xls (or *.doublehit.gene.phased.flt.gty.txt). Note The --double-hit-gene-trio-filter or --double-hit-gene-phased-filter only works for inputs specified by --vcf-file and requires parents' genotypes to infer the phases of the alleles. And sequence variants with missed parents' genotypes are ignored. Note To ignore homozygous mutation (i.e., implicating the autosomal recessive model), please add the tag --ignore-homo.
Sequence variants beyond this type of genes will be filtered out. Note --unique-gene-filter only works for inputs specified by --vcf-file.
Search the longest region (in minimal length X kb) of each interesting variant, in which there is at least one allele identical among all cases. This function can be used to highlight potential causal mutations or exclude non-disease-relevant sequence variants as the causal mutation(s) is very likely to have long shared regions among all affected family members of Mendelian dominant and recessive diseases. However, IBS checking is just a quick but over-estimation of identical by descent (IBD), which is a more accurate measure of shared regions among related individuals. KGGSeq currently has no function to estimate IBD regions but allow users to input the IBD information estimated by third-party tools into KGGSeq for annotation. Note --ibs-case-filter only works for inputs specified by --vcf-file.
User can also provide IBD (i.e., the two alleles arose from the same allele in an earlier generation) or significant linkage regions, which are estimated by a third party statistical genetics tools (such as BEAGLE, Merlin and SimWalk2). Variants within these regions will be highlighted. For an ibd or linkage annotation file, title line is needed, and the file format is like:
CHR START END
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gene database label | Supported reference genome | Description |
| refgene | hg18 and hg19 | The RefGene datasase compiled by UCSC from hg18 refGene and hg19 refGene
Note: RefSeq has NO mitochondria gene definition. |
| gencode | hg19 | The GENCODE gene sets.See home page and the paper of GENCODE for details.
Note: GECODE contains similar number of coding genes but more transcripts than RefGene. It HAS the mitochondria gene definition. |
| knowngene | hg18 and hg19 | The UCSC knonwGene datasase compiled by UCSC from hg18 knownGene and hg19 knownGene |
| ensembl | hg19 | The Ensembl gene datasase compiled by UCSC from hg19 ensGene |
With this filter, variants beyond '--gene-feature-in' region will be excluded. It is '--gene-feature-in 0,1,2,3,4,5...,15' by default.
Illustration of number codes for the gene features:| Feature | Code | Explanation |
| Frameshift | 0 | Short insertion or deletion result in a completely different translation from the original. |
| Nonframeshift | 1 | Short insertion or deletion result in loss of amino acids in the translated proteins. |
| Startloss | 2 | Indels or nucleotide substitution result in the loss of start codon(ATG) (mutated into a non-start codon). |
| Stoploss | 3 | Indels or nucleotide substitution result in the loss of stop codons (TAG, TAA, TGA) |
| Stopgain | 4 | Indels or nucleotide substitution result in the new stop codons (TAG, TAA, TGA), which may truncate the protein. |
| Splicing | 5 | variant is within 2-bp of a splicing junction (use --splicing x to change this, the unit of x is base-pair) |
| Missense | 6 | Variants result in a codon coding for a different amino acid (missense) |
| Synonymous | 7 | Nucleotide substitution does not change amino acid. |
| Exonic | 8 | Due to loss of sequences, only map a variant into exonic region |
| UTR5 | 9 | variant within a 5' untranslated region |
| UTR3 | 10 | variant within a 3' untranslated region |
| Intronic | 11 | Variants within an intron |
| Upstream | 12 | variant overlaps 1-kb region upstream of transcription start site? (use --neargene x to change this, the unit of x is base-pair) |
| Downstream | 13 | variant overlaps 1-kb region downtream of transcription end site (use --neargene x to change this, the unit of x is base-pair) |
| ncRNA | 14 | variant overlaps a transcript without coding annotation in the gene definition (see Notes below for more explanation) |
| Intergenic | 15 | variant is in intergenic region |
| Monomorphic | 16 | It is not a sequence variation actually, which may be resulted from bugs in reference genome in variant calling. |
| Unknown | 17 | Variants has no annotation. |
MostImportantFeatureGene: The gene corresponds to the variant’s smallest gene feature code.
MostImportantGeneFeature: The variant’s smallest gene feature code.
RefGeneFeatures: All matched gene features in the reference gene database.
GENCODEFeatures: All matched gene features in the GENCODE gene database.
The gene feature annotations are described in accordance with the Human Genome Variation Society (HGVS) recommendations. The followings are some examples:
KLHL17:NM_198317:c.1918A>C:p.T640P:(12Exons):exon12:missense
Means: At nucleotide 76 of gene KLHL17's transcript NM_198317, an A is changed to a C, which changes the 640th amino-acid T to be P in this transcript's protein. This transcript has 12 exons in total and this variant is in the 12th exon.
DNAJC8:NM_014280:c.237+2T>G:(9Exons):exon3GTdonor
Means:This is a T to G substitution of the 2nd nucleotide of the 2 nd intron (from its 5' part) positioned between coding DNA nucleotides 237 and 238, which will affect the GT splicing donor.
HNRNPCL2:NM_001136561:c.-169C>G:(2Exons):5UTR
Means: This is a C to G substitution of nucleotide, 169 nucleotides upstream of the ATG translation initiation codon (coding DNA -169).
PRAMEF4:NM_001009611:c.*62C>T:(4Exons):3UTR
Means: This is a C to T substitution of nucleotide coding DNA 62, located in the 3' UTR.
C1orf159:NM_017891:c.311-23T>C:(10Exons):intronic6
Means: This is a T to C substitution of the 23 nucleotide of the 6 th intron (from its 3' part) positioned between coding DNA nucleotides 310 and 311.
HNRNPCL1:NM_001013631:c.*138G>A:(2Exons):downstream
Means: This is a C to A substitution of nucleotide located 293 nucleotides downstream of the gene, i.e. the polyA-addition site.
TNFRSF18:NM_148901:c.-61C>T:(4Exons):upstream
Means: This is a C to T substitution of nucleotide, 61 nucleotides upstream of the ATG translation initiation codon (coding DNA -61).
Allele frequency trimming by 4 ways
![]() 1. Allele frequency trimming according to database: --db-filter
1. Allele frequency trimming according to database: --db-filter
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --db-filter 1kg201204,dbsnp141,ESP6500AA,ESP6500EA --rare-allele-freq 0.005 --db-filter-hard dbsnp138nf
This mode enables variants filtering according to allele frequencies recorded in public data sources. The '--db-filter' is use to set databases for filtration. In the above case, variants with allele alternative allele frequency EQUAL to or over 0.005 will be excluded (not including the boundary). No spaces are allowed between and within database names.
Moreover, '--db-filter-hard' is use to filter ALL existing variants in a database. But it should be always careful to use this hard filtering.
List of public databases for allele frequency trimming
| Genome Build | DB Label | Explanation |
| hg18 | 1kg201305 | All sample of 1000 Genomes Project release 2013 May (over 81 million sequence variants from 2504 subjects) |
| hg18 | 1kgasn2010 | 1000 Genomes Project Pilot Data SNPs + indels (2010 March call set) for ASN (asian) |
| hg18 | 1kgeur2010 | 1000 Genomes Project Pilot Data SNPs + indels (2010 March call set) for EUR (european) |
| hg18 | 1kgafr2010 | 1000 Genomes Project Pilot Data SNPs + indels (2010 March call set) for AFR (african) |
| hg18 | 1kgasn2009 | 1000 Genomes Project Pilot Data SNPs (2009 April call set) for ASN (asian) |
| hg18 | 1kgeur2009 | 1000 Genomes Project Pilot Data SNPs (2009 April call set) for EUR (european) |
| hg18 | 1kgafr2009 | 1000 Genomes Project Pilot Data SNPs (2009 April call set) for AFR (african) |
| hg18 | dbsnp137 | dbSNP version 137,compiled from UCSC with coordinates converted by liftover. |
| hg18 | dbsnp138 | dbSNP version 138, compiled from UCSC with coordinates converted by liftover. |
| hg18 | dbsnp138nf | dbSNP version 138 without the flagged SNPs by UCSC (so nonflagged: nf). In UCSC the flagged SNPs include SNPs clinically associated by dbSNP, mapped to a single location in the reference genome assembly, and not known to have a minor allele frequency of at least 1%. The coordinates were converted from hg19 to hg18 by liftover. |
| hg18 | dbsnp141 | dbSNP version 141, compiled from NCBI dbSNP V141. Note Indels are ingored. |
| hg18 | ESP5400 | a public variants dataset from NHLBI GO Exome Sequencing Project (ESP) |
| hg19 | 1kg201305 | All sample of 1000 Genomes Project release 2013 May (over 81 million sequence variants from 2504 subjects) |
| hg19 | 1kg201204 | All sample of 1000 Genomes Project release 2012 April |
| hg19 | 1kgafr201204 | African descendent samples of 1000 Genomes Project release in 2012 April |
| hg19 | 1kgeur201204 | European descendent samples of 1000 Genomes Project release in 2012 April |
| hg19 | 1kgamr201204 | American descendent samples of 1000 Genomes Project release in 2012 April |
| hg19 | 1kgasn201204 | Asian descendent samples of 1000 Genomes Project release in 2012 April |
| hg19 | dbsnp135 | dbSNP version 135, compiled from UCSC. |
| hg19 | dbsnp137 | dbSNP version 137, compiled from UCSC. |
| hg19 | dbsnp138 | dbSNP version 138, compiled from UCSC. |
| hg19 | dbsnp138nf | dbSNP version 138 without the flagged SNPs by UCSC (so nonflagged: nf). In UCSC the flagged SNPs include SNPs clinically associated by dbSNP, mapped to a single location in the reference genome assembly, and not known to have a minor allele frequency of at least 1%. |
| hg19 | dbsnp141 | dbSNP version 141, compiled from NCBI dbSNP V141. Note Indels are ingored. |
| hg19 | ESP5400 | a public variants dataset from NHLBI GO Exome Sequencing Project (ESP) |
| hg19 | ESP6500AA | a public variants dataset of African American from NHLBI GO Exome Sequencing Project (ESP) |
| hg19 | ESP6500EA | a public variants dataset of European American from NHLBI GO Exome Sequencing Project (ESP) |
| hg19 | exac | Exome Aggregation Consortium (ExAC): Variants from 61,486 unrelated individuals sequenced as part of various disease-specific and population genetic studies (ESP) |
MaxDBAltAF: The maximum alternative allele frequency across datasets specified.
altFreq@hgXX_XXXXX: The alternative allele frequency at a dataset hgXX_XXXXX.
NoteVariants without allele frequencies in reference databases are marked by '.' in the results. Moreover, Variants not EXISTING in reference database are marked by 'N'.
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --local-filter path/to/file3 --rare-allele-freq 0.005 --local-filter-hard path/to/file4
The '--local-filter' option sets local datasets for filterating. KGGSeq accepts local dataset files in the following format: each file has 5 columns [Chromosome, Physical Position, Reference Allele, Alternative Allele(s), and Frequency of alternative allele(s)], separated by tab character. KGGSeq provides a function to convert data in VCF format to that in the local dataset format:
java -jar ./kggseq.jar --make-filter --vcf-file path/to/file --buildver-in hg18 --out test.hg18.var --buildver-out hg19
An example file is like:
Chrom Position Reference Alternative Freq
1 469 C G 0.150
1 492 C T 0.175
1 519 G C 0.067
1 874290 - 0ACAGAG 0.809
1 875913 CAG 3 0.8431
Note The title line is needed and Freq is optional.
![]() 3. An in-house VCF file can be used for allele frequency trimming: --local-filter-vcf
3. An in-house VCF file can be used for allele frequency trimming: --local-filter-vcf
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --local-filter-vcf path/to/file3 --rare-allele-freq 0.005 --local-filter-vcf-hard path/to/file4
If the data were stored in different files chromosome by chromosome, you can use ' _CHROM_' to denote the chromosome names [1...Y] or directly specify [1,2,X] in the file name so that kggseq can read data in multiple files in a run.
Similarly, '--local-vcf-filter-hard' is use to filter ALL existing variants in a database.![]() 4. An in-house VCF file without genotypes can also be used for allele frequency trimming: --local-filter-no-gty-vcf
4. An in-house VCF file without genotypes can also be used for allele frequency trimming: --local-filter-no-gty-vcf
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --local-filter-no-gty-vcf path/to/file3 --rare-allele-freq 0.005 --local-filter-no-gty-vcf-hard path/to/file4
Note1. To remove variants in database regardless of their frequencies, please use '--rare-allele-freq 0'.
Note2. To keep variants in a frequency range,[a,b], please use '--allele-freq a,b' option. The boundaries a and b are included.
Note3. The options, '--allele-freq' and '--rare-allele-freq', are mutually exclusive and the latter has higher priority.
Deleteriousness prediction annotation
Prioritize according to functional impact scores: --db-score
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --db-score dbnsfp
Annotate the non-synonymous sequence variants (i.e., those alter proteins) by publicly avaiable functional impact scores. Currently KGGSeq only support the dataset of dbNSFP, an integrated database of multiple algorithms for the comprehensive collection of human of deleteriousness/functional predictions at non-synonymous sequence variants of human genome. These scores will finally be combined by Logistic regression method to estimate the posterior probability of being a pathogenic variants. And the --filter-nondisease-variant can be used to jointly filterd out variant predicted to be non-disease causal variant.
Variant type, region and gene filtering
![]() Ingore insertion and deletion sequence variants (indels): --ignore-indel
Ingore insertion and deletion sequence variants (indels): --ignore-indel
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --ignore-indel
![]() Accordingly, users can also ask kggseq to ingore Single nucleotide variants (SNVs) : --ignore-snv
Accordingly, users can also ask kggseq to ingore Single nucleotide variants (SNVs) : --ignore-snv
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --ignore-snv
By default, KGGSeq will process both SNV and Indel.
![]() Ignore some genomic regions: --regions-out
Ignore some genomic regions: --regions-out
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --regions-out chrX,chrY:1-10000
![]() Alternatively, one can also exclusively process sequence variants at some genome regions:
Alternatively, one can also exclusively process sequence variants at some genome regions:
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --regions-in chr2,chr4:21212-233454
![]() Ignore variants within some genes: --genes-out
Ignore variants within some genes: --genes-out
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --genes-out TCF4,CNNM2,ANK3
![]() Alternatively, one can KEEP variants only within some genes:
Alternatively, one can KEEP variants only within some genes:
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --genes-in TCF4,CNNM2,ANK3
Super duplicate region filtering
![]() Filter out variants in super duplicate regions: --superdup-filter
Filter out variants in super duplicate regions: --superdup-filter
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --superdup-filter
KGGSeq can eliminate sequence variants within putative super-duplicate genomic regions defined in a dataset (genomicSuperDups) from UCSC datasets , which have higher genotyping error rate. (See more ).
Note Anoher option --superdup-annot allows you to just mark (but not to remove) the variants in the super duplicate regions. A column named SuperDupKValue will be added in the resulting dataset
Too-many-variant gene filtering
![]() Filter out genes with too many putative pathogenic variants: --gene-var-filter
Filter out genes with too many putative pathogenic variants: --gene-var-filter
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 [options to filter out common and neutral variants] --gene-var-filter 4
After a number of hard-filtering by allele frequencies and pathogenic prediction, you may see some genes harboring candidate variants. Empirically, these genes are usually not interesting and their mutations are often produced by some unknown factors such pseudogene or platform bias. As a rule of thumb, it is safe to set a cutoff 4 or more.
Phenotype-relevant filtering
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --filter-model case-unique
Case unique: extract variants with alternative alleles only present among affected individuals.Control unique: extract variants with alternative alleles only present among unaffected individuals.
Note The --filter-model only works for inputs specified by --vcf-file and --v-assoc-file.
Note This function can work with options to flexibly set the sample size and missingness of genotypes, detalied in the Variant QC part.
![]() Filtration by statistical association: --filter-model association
Filtration by statistical association: --filter-model association
java -jar ./kggseq.jar --buildver hg19 --filter-model association --v-assoc-file path/to/file3 --p-value-cutoff 0.01 --multiple-testing benfdr --qqplot Exclude variants which are not interesting in terms of statistical p-values for association calculated by PLINK/Seq . The '--p-value-cutoff' sets p-value cutoffs for filtration; the '--multiple-testing' sets the approaches for multiple testing comparisons; and the '--qqplot' enables Q-Q plot of the association results.
| Multiple testing method | Description |
| no | No multiple testing methods. Set a fixed p value cutoff by --p-value-cutoff. |
| benfdr | Benjamini-Hochberg method, which aims to control the FDR (specified by --p-value-cutoff), or "False Discovery Rate," rather than the FWE or "FamilyWise Error rate". |
| bonhol | Bonferroni-Holm method, which is less conservative and more powerful than the Standard Bonferroni method given a family-wise error rate specified by --p-value-cutoff. |
| bonf | Standard Bonferroni correction given a family-wise error rate specified by --p-value-cutoff. |
Note This functions also supports inputs specified by --vcf-file, --assoc-file, which is the output of PLINK/Seq gene-based association analysis result, and --vaast-simple-file, which is the output of VAAST gene-based association analysis result.
Annotation
Functional annotation
Provide genomic functional annotation of variants: --genome-annot
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --genome-annot
Currently supported functional annotation categories are Pseudogenes, transcription factor binding sites (TFBSs), enhancers, and UniProt Feature. The option will append three extra fields to the output file:
Pseudogenes: Pseudogenes listed in http://tables.pseudogene.org/set.py?id=Human61
DispensableDeleteriousVariants: Number of dispensable deleterious variants for the gene
in the 1000 Genomes Project
TFBSconsSite[name:rawScore:zScore]: Conserved TFBSs in the UCSC genome browser
vistaEnhancer[name:+ve/-ve]: Known enhancers in the VISTA enhancer browser
UniProtFeature: Annotate a variant of coding gene using the UniProt protein annotations.
| UniProtFeature | Definition |
| region of interest | Extent of a region of interest in the sequence |
| active site | Amino acid(s) involved in the activity of an enzyme. |
| calcium-binding region | Extent of a calcium-binding region. |
| glycosylation site | Glycosylation site. |
| chain | Extent of a polypeptide chain in the mature protein. |
| coiled-coil region | Extent of a coiled-coil region |
| compositionally biased region | Extent of a compositionally biased region |
| sequence conflict | Different papers report differing sequences. |
| cross-link | Posttranslationally formed amino acid bonds. |
| disulfide bond | Disulfide bond. |
| DNA-binding region | Extent of a DNA-binding region. |
| domain | Extent of a domain, which is defined as a specific |
| helix | DSSP secondary structure |
| intramembrane region | |
| initiator methionine | Initiator methionine. |
| modified residue | |
| lipid moiety-binding region | |
| metal ion-binding site | Binding site for a metal ion. |
| short sequence motif | Short (up to 20 amino acids) sequence motif of biological interest |
| mutagenesis site | Site which has been experimentally altered. |
| non-consecutive residues | Non-consecutive residues. |
| non-standard amino acid | Non-standard aminoacid used for pyrrolysine |
| non-terminal residue | |
| nucleotide phosphate-binding region | Extent of a nucleotide phosphate binding region. |
| peptide | Extent of a released active peptide. |
| propeptide | Extent of a propeptide. |
| repeat | Extent of an internal sequence repetition. |
| signal peptide | Extent of a signal sequence (prepeptide). |
| site | Any interesting single amino-acid site on the sequence. |
| strand | DSSP secondary structure |
| transit peptide | |
| topological domain | Topological domain |
| transmembrane region | Extent of a transmembrane region. |
| turn | DSSP secondary structure |
| unsure residue | Uncertainties in the sequence |
| sequence variant | Authors report that sequence variants exist. |
| splice variant | Description of sequence variants produced by alternative |
| zinc finger region | Extent of a zinc finger region. |
| binding site |
Note Users should be aware of possible mapping error due to these pseudogenes.
Shared protein-protein interactions (PPIs)
Examine whether variants share the same PPIs with some genes of interest: --candi-list gene1,gene2,...,geneN --ppi-annot ppiDatabase
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --candi-list gene1,gene2,...,geneN --ppi-annot string --ppi-depth 1
Note Currently, the only supported PPI database is STRING (i.e., string).
Note Instead of specifying the gene list using the --candi-list option, users can specify a file containing the list using the --candi-file option.
Note The maximum distance of a PPI between a specifified candidate genes and a gene containing the potential causal variants can be adjusted using the --ppi-depth option. The defaul is --ppi-depth 1.
IsWithinCandidateGene: Whether the variant is within a candidate gene
PPI: PPI shared by at least one gene of interest and the gene
containing the variant
Shared pathways
Examine whether variants share the same pathways with some genes of interest: --candi-list gene1,gene2,...,geneN --pathway-annot pathwayDatabase
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --candi-list gene1,gene2,...,geneN --pathway-annot PathwayDatabase
Currently, users can choose between 5 pathway databases from GSEA. The following are brief descriptions of the 5 datasets COPIED from the page of GSEA.
| PathwayDatabase | Description | Version |
|---|---|---|
| cano | Gene sets from the pathway databases. Usually, these gene sets are canonical representations of a biological process compiled by domain experts. (1452 gene sets) | MSigDB 3.1 |
| cura | Curated gene sets: Gene sets collected from various sources such as online pathway databases, publications in PubMed, and knowledge of domain experts. The gene set page for each gene set lists its source. (4850 gene sets) | MSigDB 3.1 |
| onco | Oncogenic signatures: Gene sets represent signatures of cellular pathways which are often dis-regulated in cancer. The majority of signatures were generated directly from microarray data from NCBI GEO or from internal unpublished profiling experiments which involved perturbation of known cancer genes. In addition, a small number of oncogenic signatures were curated from scientific publications. (189 gene sets) | MSigDB 3.1 |
| cmop | Computational gene sets: Computational gene sets defined by mining large collections of cancer-oriented microarray data. (858 gene sets) | MSigDB 3.1 |
| onto | GO gene sets Gene sets are named by GO term and contain genes annotated by that term. GSEA users: Gene set enrichment analysis identifies gene sets consisting of co-regulated genes; GO gene sets are based on ontologies and do not necessarily comprise co-regulated genes. (1454 gene sets) | MSigDB 3.1 |
Note The --candi-list and --candi-file options can be shared with --ppi-annot.
The option will append the following fields to the output file:
IsWithinCandidateGene: Whether the variant is within a candidate gene
SharedPathway: Pathway shared by at least one gene of interest and the gene
containing the variant
Text mining
PubMed text mining:--pubmed-mining searchTerm or --pubmed-mining-gene searchTerm
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --pubmed-mining search+Term1,search+Term2
KGGSeq can mine the titles and abstracts of published papers in PubMed via the NCBI E-utilities to find a co-mention with the searched term(s) of interest (i.e., disease) and the genes/cytogeneic regions in which the
variants are located. The option --pubmed-mining will append the following two fields to the output file while the --pubmed-mining-gene will only add the PubMedIDGene information.
PubMedIDIdeogram : PubMed ID of articles in which the term and the cytogeneic
position of the variant are co-mentioned
PubMedIDGene : PubMed ID of articles in which the term and the gene
containing the variant are co-mentioned
IMPORTANT Please use + instead of blank characters within a search term.
OMIM term annotation:--omim-annot
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --omim-annot
KGGSeq can extract all available disorder names linked to a gene in which the
variants are located from the OMIM dataset morbidmap. The command will append two extra fields to the output file:
DiseaseName(s)MIMid : Disorder, <disorder MIM no.> (<phene mapping key>)
Phenotype mapping method <phene mapping key>:
1 - the disorder is placed on the map based on its association with
a gene, but the underlying defect is not known.
2 - the disorder has been placed on the map by linkage; no mutation has
been found.
3 - the molecular basis for the disorder is known; a mutation has been
found in the gene.
4 - a contiguous gene deletion or duplication syndrome, multiple genes
are deleted or duplicated causing the phenotype.
GeneMIMid : Gene/locus MIM no.
COSMIC canncer information: --cosmic-annot
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --indiv-pair NONTUMOR.1:TUMOR.1,NONTUMOR.2:TUMOR.2 --genotype-filter 8 --cosmic-annot
KGGSeq can retrieve the confirmed somatic mutation information registered for variants and genes in the COSMIC database.
The command will append a field to the output file. e.g.,
COSMICCancerInfo : known cancer and occurrence time of the somatic mutation in COSMIC dataset. e.g., {serous_carcinoma=3, astrocytoma_Grade_IV=1}
This somatic mutation occurred 3 times in samples of 'serous_carcinoma' and once in samples of 'astrocytoma_Grade_IV'.
Prediction
One attractive feature of KGGSeq is that it combines the functional impact scores from multiple algorithms (i.e., SIFT and PolyPhen2) to predict whether variants are disease-causing or not. If you use this feature in any published work, please cite the following manuscript:Li et al. Predicting Mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013 Jan;9(1):e1003143
Predicting Mendelian disease-causing variants
Predict Mendelian disease-causing variants: --db-score dbnsfp --mendel-causing-predict
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --db-score dbnsfp --mendel-causing-predict all --filter-nondisease-variant
Use at most 12 available functional impact scores (listed below) collected in dbNSFP database to RE-predict whether a nonsynonymous single nucleotide variant (SNV) will potentially be Mendelian disease causal or not by Logistic regression model (See methods in our paper, Li et al. PLoS Genet. 2013 Jan;9(1):e1003143). By default, KGGSeq tries all subsets of the scores (at least two) for a combinatorial prediction and finally present a prediction which can pass the threshold to classify the SNV as a potentially disease causal or no. In conjunction with the prediction model, the --filter-nondisease-variant tag will be used to filter out the nondisease-causative variants predicted by Logistic regression model (See methods in Li et al. PLoS Genet. 2013 Jan;9(1):e1003143). .Note that this trying will increase the false positive rate although it decreases the false negative rate. So you may need more information to justify the involved gene(s).
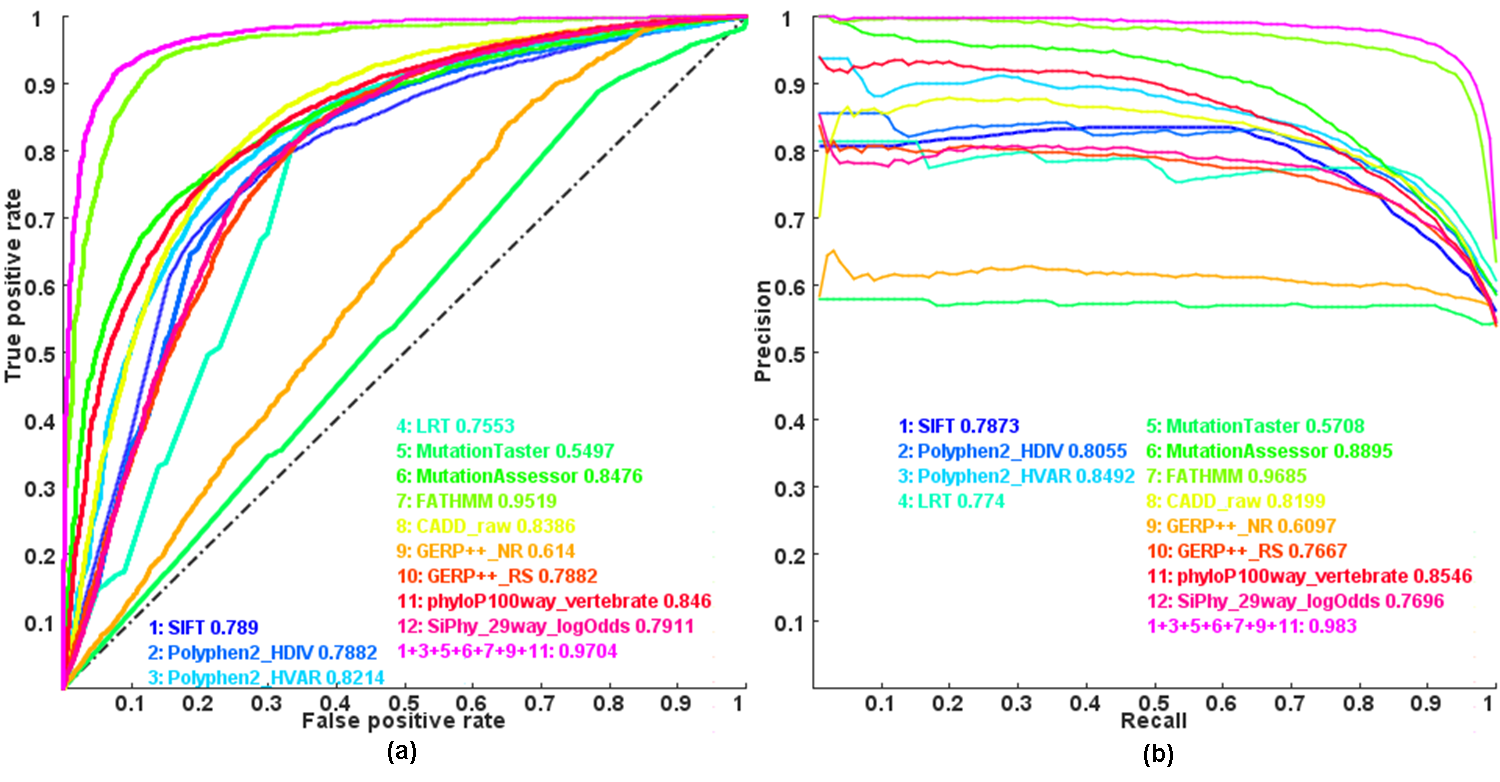
Figure: Receiver operating characteristic (ROC) and Precision recall(PR) curves and area under the curves (AUC) of individual scores and combined score by Logistic regression model
Note: this is an updated plot of Figure 1 in our paper . Here we combined more individual functional impact scores, which is the only difference.
On the other hand, one can FIX the prediction using a specified subset or full set of the 5 impact scores by option like --mendel-causing-predict 4,6,7
The coding for the functional impact scores used in'--mendel-causing-predict'options is listed below:
| Coding | Method | Description (Mostly copied from dbNSFP) |
| - | SLR | Sitewise Likelihood-ratio (SLR) test statistic for testing natural selection on codons. A negative value indicates negative selection, and a positive value indicates positive selection. Larger magnitude of the value suggests stronger evidence. |
| 1 | SIFT | SIFT uses the 'Sorting Tolerant From Intolerant' (SIFT) algorithm to predict whether a single amino acid substitution affects protein function or not, based on the assumption that important amino acids in a protein sequence should be conserved throughout evolution and substitutions at highly conserved sites are expected to affect protein function.A small scoreindicates a high chance for a substitutionto damage the protein function. |
| 2 | Polyphen2_HDIV | Polyphen2 score based on HumDiv, i.e. hdiv_prob. The score ranges from 0 to 1, and the corresponding prediction is "probably damaging" if it is in [0.957,1]; "possibly damaging" if it is in [0.453,0.956]; "benign" if it is in [0,0.452]. Score cutoff for binary classification is 0.5, i.e. the prediction is "neutral" if the score is smaller than 0.5 and "deleterious" if the score is larger than 0.5. Multiple entries separated by ";" |
| 3 | Polyphen2_HVAR | Polyphen2 predicts the possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations by an iterative greedy algorithm. In the present study, we use the original scores generated by the HumVar (instead ofHumDiv) trained model as it is preferred for the diagnosis of Mendelian diseases. The scores range from 0 to 1. A substitution with larger score has a higher possibility to damage the protein function. |
| 4 | LRT | LRT employed a likelihood ratio test to assess variant deleteriousnessbased on a comparative genomics data set of 32 vertebrate species. The identified deleterious mutations could disrupt highly conserved amino acids within protein-coding sequences, which are likely to be unconditionally deleterious.The scores range from 0 to 1. A larger score indicates a larger deleterious effect. |
| 5 | MutationTaster | MutationTaster assesses the impact of the disease-causing potential of a sequence variant by a naive Bayes classifier using multiple resources such as evolutionary conservation, splice-site changes, loss of protein features and changes that might affect mRNA level. The scores range from 0 to 1. The larger score suggests a higher probability to cause a human disease. |
| 6 | MutationAssessor | MutationAssessor "functional impact of a variant : predicted functional (high, medium), predicted non-functional (low, neutral)" Please refer to Reva et al. Nucl. Acids Res. (2011) 39(17):e118 for details |
| 7 | FATHMM | FATHMM default score (weighted for human inherited-disease mutations with Disease Ontology); If a score is smaller than -1.5 the corresponding NS is predicted as "D(AMAGING)"; otherwise it is predicted as "T(OLERATED)". If there's more than one scores associated with the same NS due to isoforms, the smallest score (most damaging) was used. Please refer to Shihab et al Hum. Mut. (2013) 34(1):57-65 for details |
| 8 | VEST3 | VEST 3.0 score. Score ranges from 0 to 1. The larger the score the more likely the mutation may cause functional change. In case there are multiple scores for the same variant, the largest score (most damaging) is presented. Please refer to Carter et al., (2013) BMC Genomics. 14(3) 1-16 for details. Please note this score is free for non-commercial use. For more details please refer to http://wiki.chasmsoftware.org/index.php/SoftwareLicense. Commercial users should contact the Johns Hopkins Technology Transfer office. |
| 9 | CADD_score | Combined Annotation Dependent Depletion (CADD) score for funtional prediction of a SNP. Please refer to Kircher et al. (2014) Nature Genetics 46(3):310-5 for details. The larger the score the more likely the SNP has damaging effect. |
| 10 | GERP++_NR | Neutral rate |
| 11 | GERP++_RS | RS score, the larger the score, the more conserved the site |
| 12 | PhyloP100way_vertebrate | PhyloP estimates the evolutional conservation at each variant from multiple alignments of 100 vertebrate genomes to the human genome based on a phylogenetic hidden Markov model. |
| 13 | 29way_logOdds | SiPhy score based on 29 mammals genomes. The larger the score, the more conserved the site. |
The option will append the following fields to the output file:
...
Polyphen2_HDIV_pred: Polyphen2 prediction based on HumDiv, "D" ("porobably damaging"),
"P" ("possibly damaging") and "B" ("benign"). Multiple entries separated by ";"
Polyphen2_HVAR_pred: Polyphen2 prediction based on HumVar, "D" ("porobably damaging"),
"P" ("possibly damaging") and "B" ("benign"). Multiple entries separated by ";"
LRT_pred: Classification using LRT (D = deleterious, N = neutral,
or U = unknown)
MutationTaster_pred: Classification using MutationTaster (A =
disease_causing_automatic, D = disease_causing, N =
polymorphism, or P = polymorphism_automatic)
MutationAssessor_pred: MutationAssessor "functional impact of a variant :
predicted functional (high, medium), predicted non-functional (low, neutral)"
DiseaseCausalProb_ExoVarTrainedModel: Conditional probability of being Mendelian disease-causing
given the above prediction scores under a logistic regression
model trained by our dataset ExoVar.
IsRareDiseaseCausal_ExoVarTrainedModel: Classification using the logistic regression model
(Y = disease-causing or N = neutral)
BestCombinedTools:OptimalCutoff:TP:TN : The subset of original prediction tools (out of the 13 tools) used for the combined prediction by our Logistic Regression model which have the largest posterior probability among all possible combinatorial subsets: the cutoff leads to the maximal Matthews correlation coefficient (MCC): the corresponding true positive and true negative at the maximal MCC.
Note By default the option --mendel-causing-predict all is always switched on unless other predicion types are specified.
Predicting somatic driver mutations of cancers
Predict the driver somatic-mutations and genes of cancers by an integrative methods: --db-score dbnsfp --cancer-driver-predict
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --indiv-pair NONTUMOR.1:TUMOR.1,NONTUMOR.2:TUMOR.2 --genotype-filter 8 --cosmic-annot --db-score dbnsfp --cancer-driver-predict all --filter-nondisease-variant
A driver mutation is defined as the mutation that leads to selective advantage to a cancer clone by either increasing the cancer cells' survival or reproduction. Use at most 12 available functional impact scores (listed above, exept for LRT_Omega) collected in dbNSFP database to RE-predict whether a nonsynonymous single nucleotide variant (SNV) will potentially be the somatic driver mutations or not by Logistic regression model trained under COSMIC database. By default, KGGSeq tries all subsets of the scores (at least two) for a combinatorial prediction and finally present a prediction which can pass the threshold to classify the SNV as a potentially disease causal or no. In conjunction with the prediction model, the --filter-nondisease-variant tag will be used to filter out the nondisease-causative variants predicted by Logistic regression model.
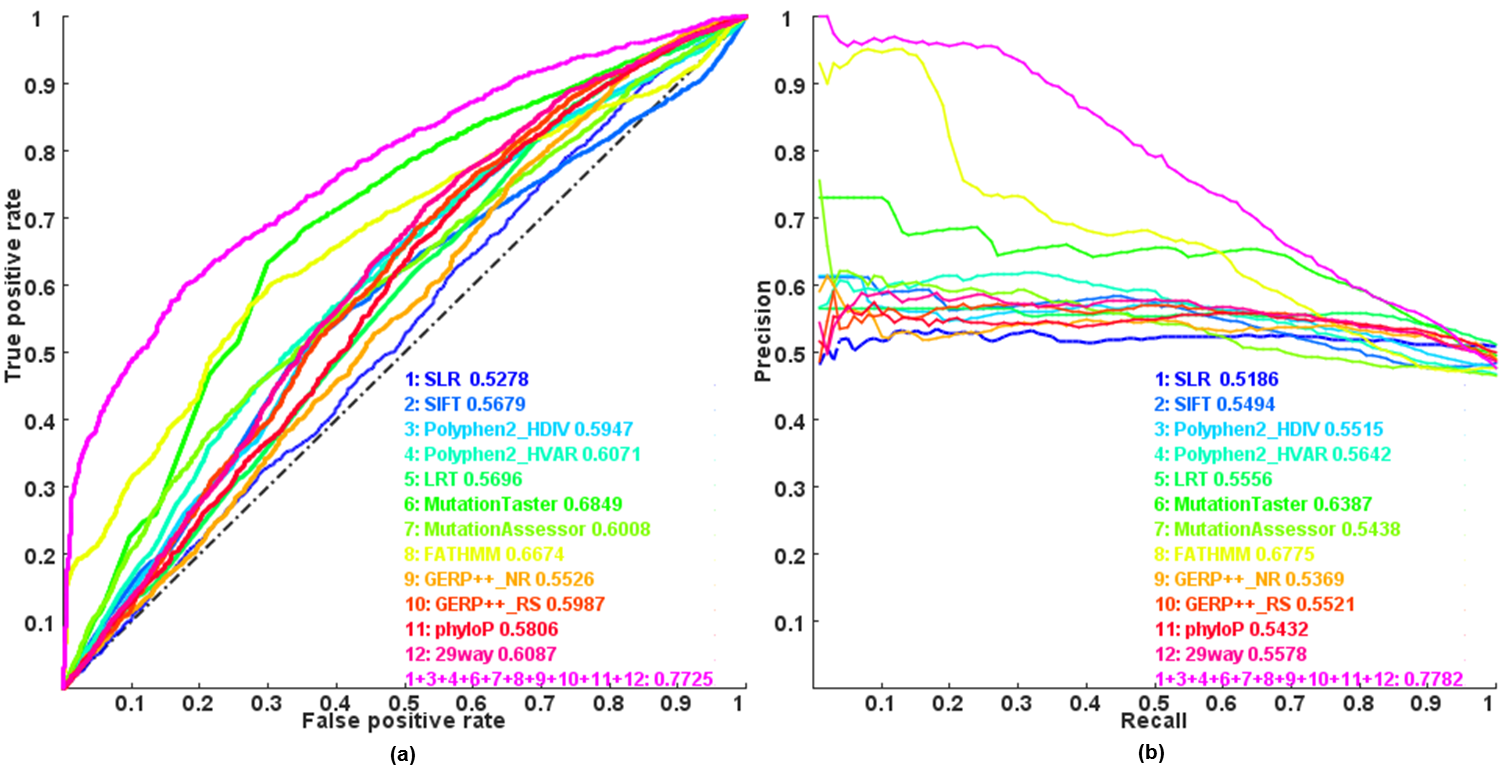
Figure: Receiver operating characteristic (ROC) and Precision recall(PR) curves and area under the curves (AUC) of individual scores and combined score by Logistic regression model
The option will append the following fields to the output file:
...
CancerDriverProb_COSMICTrainedModel: Conditional probability of being cancer driver mutation
given the above prediction scores under a logistic regression
model trained by the COSMIC dataset.
IsCancerDriver_COSMICTrainedModel: Classification using the logistic regression model
(Y = cancer-drivering or N = passenger)
BestCombinedTools:OptimalCutoff:TP:TN: The subset of original prediction tools (out of the 13 tools) used for the combined prediction by our Logistic Regression model which have the largest posterior probability among all possible combinatorial subsets: the cutoff leads to the maximal Matthews correlation coefficient (MCC): the corresponding true positive and true negative at the maximal MCC.
In addition, genes of these predicted cancer-driver somatic mutations are evaluated by our statistical approach (FUSE).
Kwan JS, Ng OL, Sham PC and Li MX. FUSE: FUnctional and Statistical Evidence combined to identify driver genes in cancer genomes. (Submitted)
In the output file of the cancer-driver genes (*.cancer.gene.xls), there are detailed statistical evaluation results of each gene:
GeneSymbol: Official gene symbols defined by HGNC.
AvgCodingLen: The actual or averaged (of multiple isoforms) length (in kb) of all exons in a gene.
#AllNS: The observed number of non-synonymous and splicing somatic sequence variants.
#PositiveNS: The number of predicted possible cancer-driver non-synonymous and splicing somatic sequence variants.
#S: The observed number of synonymous somatic sequence variants.
CancerDriverPValue: The probability of having the observed number or more predicted possible cancer-driver mutations in a gene by chance.
NonCancerDriverPValue: The probability of having the observed number or more synonymous mutations in a gene. The synonymous somatic mutations are assumed to not contribute to the cancer cell growth advantage. This measure can be used to exclude a gene having synonymous somatic mutations due to technique artifacts or mapping errors.
Reads: The reads carrying reference and alternative alleles between tumor and non-tumor tissues. Example:
NORMAL.107 <-> TUMOR.107,112/0,85/32,5.00E-11,84.329414|NORMAL.105 <-> TUMOR.105,52/0,46/24,2.48E-7,54.260868
Here the somatic mutations are observed in two subjects separated by |. For the first subject, the reads carrying reference and alternative alleles between tumor and non-tumor tissues are 112/0 and 85/32 respectively, which are significantly different between the two different tissues, p=5.00E-11 and odds ratio =84.33.
PPI: protein-protein interactions between this gene and inputted disease genes.
Pathway: biological pathways shared by this gene and inputted disease genes.
PubMedID: PubMed ID of articles in which the disease name and the gene are co-mentioned.
COSMICCancerInfo: known cancer and occurrence time of the somatic mutations of this gene in COSMIC dataset.
Similarly, the FUSE method is also used to statistically assess the biological pathways enriched in predicted cancer-driver somatic mutations: --db-score dbnsfp --cancer-driver-predict --pathway-db pathwayDatabase or --pathway-db path/to/mypahway/file
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --indiv-pair NONTUMOR.1:TUMOR.1,NONTUMOR.2:TUMOR.2 --genotype-filter 8 --cosmic-annot --db-score dbnsfp --cancer-driver-predict all --filter-nondisease-variant --pathway-db pathwayDatabase
Currently, there are 5 pathway databases from GSEA. The following are brief descriptions of the 5 datasets COPIED from the page of GSEA.
| PathwayDatabase | Description | Version |
|---|---|---|
| cano | Gene sets from the pathway databases. Usually, these gene sets are canonical representations of a biological process compiled by domain experts. (1452 gene sets) | MSigDB 3.1 |
| cura | Curated gene sets: Gene sets collected from various sources such as online pathway databases, publications in PubMed, and knowledge of domain experts. The gene set page for each gene set lists its source. (4850 gene sets) | MSigDB 3.1 |
| onco | Oncogenic signatures: Gene sets represent signatures of cellular pathways which are often dis-regulated in cancer. The majority of signatures were generated directly from microarray data from NCBI GEO or from internal unpublished profiling experiments which involved perturbation of known cancer genes. In addition, a small number of oncogenic signatures were curated from scientific publications. (189 gene sets) | MSigDB 3.1 |
| cmop | Computational gene sets: Computational gene sets defined by mining large collections of cancer-oriented microarray data. (858 gene sets) | MSigDB 3.1 |
| onto | GO gene sets Gene sets are named by GO term and contain genes annotated by that term. GSEA users: Gene set enrichment analysis identifies gene sets consisting of co-regulated genes; GO gene sets are based on ontologies and do not necessarily comprise co-regulated genes. (1454 gene sets) | MSigDB 3.1 |
Alternatively, uses can specify the own pathway gene sets in a text file. The format of the pathway file is:
[Column 1: Pathway ID]
[Column 2: Pathway URL]
[Column 3..N: Gene Symbols in the pathway; separated by spaces].
No title row is required!!!
e.g.,
----------------------------------------------
KEGG_GLYCOLYSIS_GLUCONEOGENESIS http://www.broadinstitute.org/KEGG_GLYCOLYSIS_GLUCONEOGENESIS.html LDHC LDHB LDHA
KEGG_CITRATE_CYCLE_TCA_CYCLE http://www.broadinstitute.org/KEGG_CITRATE_CYCLE_TCA_CYCLE.html GJB1 OGDHL OGDH PDHB IDH3G LDHB
...
...
In the output file of the cancer-driver pathways (*.cancer.pathway.xls), there are detailed statistical evaluation results of each pathway:
Pathway: the input pathway name or id
Size: the number of genes within the pathway
TotalExonLen: the length (in kb)of exons of all genes in the pathway
TotalNSSomatNum: the number of non-synonymous and splicing somatic sequence variants of all genes in the pathway
TotalSSomatNum: the number of synonymous somatic sequence variants Of all genes in the pathway
Genes: genes with predicted possible cancer-driver non-synonymous and splicing somatic sequence variants
CancerDriverPValue: The probability of having the observed number or more predicted possible cancer-driver mutations within a pathway by chance.
NonCancerDriverPValue: The probability of having the observed number or more synonymous mutations within a pathway by chance. The synonymous somatic mutations are assumed to not contribute to the cancer cell growth advantage. This measure can be used to exclude a pathway having synonymous somatic mutations due to technique artifacts or mapping errors.
HypergeometricPValue: Hypergeometric enrichment set test p-value of a biological pathway enriched by genes with promising cancer-driver gene p-values (described above). The selection of the genes for hypergeometric test is determined by Benjamini Hochberg FDR test with a cutoff specified by --pathway-gene-p.
Predicting complex disease-causing variants
Note This feature will be available soon. Stay tuned for an update!
Plotting
Generate a histogram of minor allele frequency (MAF) --mafplot
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --db-filter hg18_1kgasn2010,hg18_1kgeur2010,hg18_1kgafr2010,hg18_ESP5400 --allele-freq 0,1 --mafplot
Note Currently, --mafplot only supports the inputs specified by --vcf-file and --v-assoc-file.
Generate a Quantile-Quantile (Q-Q) plot for association p-values --qqplot
java -jar kggseq.jar --v-assoc-file path/to/file --out path/to/prefixname --qqplot
Note Currently, --mafplot only supports the inputs specified by --assoc-file, --v-assoc-file.
FAQ?
1) Does Kggseq provide gene expression information in the output?
Kggseq does not implement any gene expression database for annotating location or quantity of gene expression. But users can input Kggseq’s final gene list to GeneCards for gene expression information.
2) Why Kggseq does not read my VCF file?
If you use standard VCF output from GATK pipeline, it usually contains variants on mitochondrial DNA. However, mitochondrial DNA is not annotated by gene feature database. Therefore Kggseq currently only accept VCF file excluding variants on ChrM.
3) Is there a link for annotating shared pathways in terms of biological function?
Kggseq implements MsigDB database, therefore each pathway item output by Kggseq can be directed to a weblink which contain concrete information for the corresponding pathway.
4) How shall I prepare the candidate gene list? What is the gene list for?
The candidate gene list can include any gene interesting to the user. It may be verified causal gene for the disease, or potential causal gene. The gene list is used to fish more hidden interesting genes in order to narrow down the searching region. If no gene list provided, Kggseq can still perform routine ‘filtration + sharing’ strategy for sequencing data.
5)Suppose I have in-house control genomes in VCF format, how can I transform it into Kggseq acceptable file?
There are two alternative ways to allow you use the in-house VCF data (containing genotypes of multiple individuals) as filter.
1. Convert the VCF data into a standard kggseq local filter format by the command: --make-filter --vcf-file path/to/vcffile --buildver-in hg18 --out test.hg19.var --buildver-out hg19 first. And then set the --local-filter test.hg19.var
2. Directly set --local-filter-vcf path/to/vcffile. However, this way is less efficient than the above way as it will take additional time to parse the VCF data.
6) What is the default setting for minor allele frequency (MAF) cut-offs in the filtration step? Is there a practical guidance for MAF selection?
The default setting for MAF cut-offs is 0, which means only novel variants can survive in the filtration process. MAF selection is a difficult problem, but generally two suggestions can be adopted: for rare Mendelian disease, MAF cut-off at 0 or 0.01 is reasonable; for common complex disease of relatively rare alleles with large effect size, MAF cut-off is dependent on disease prevalence, penetrance and genetic model, so a higher cut-off like 0.05 might be more reasonable.
7) Can I use ANNOVAR and Kggseq together on my dataset?
Kggseq is quite flexible for interacting with other sequence-oriented analytical programs/software (including SOAPSNP, ANNOVAR, PLINK/seq, VAAST, etc). It can accept various input formats, and output different kinds of sequence data. In case of ANNOVAR, Kggseq can read ANNOVAR-formatted sequence variants, and write the final remaining variants in ANNOVAR format.
8)Can I run Kggseq on my Lenovo or Mac laptop? Is it time consuming to run a complete Kggseq process?
Normally, Kggseq run well and fast with >=1 GB RAM memory. Hence current laptop are certainty affordable for running Kggseq. The whole process need only <10 minutes, unless PubMed searching is set in the parameter file. For PubMed searching, it really depends on the speed of the network and also the loading of the NCBI PubMed database.
9)How do I report an error or bugs to Kggseq?
You are welcomed to join our google group (http://groups.google.com/group/kggseq_user?hl=en). This site is used for communication and discussion of Kggseq usage and functions. You can also directly write an email to limx54@yahoo.com, or kggseq_user@googlegroups.com.